 2016, Vol. 35
2016, Vol. 35

近十几年来,高通量测序技术的快速发展,极大地推动了人们对各个物种特别是人类基因组序列的深入了解。在对基因的研究过程中,基因组上的结构变异在进化与自然变异中的有着重要意义[1-4],其中拷贝数变异(CNV)由于在人类的某些重大疾病中扮演重要角色受到研究者们的重视[5-8]。因此,对识别CNV的策略和方法的研究相对于检测其他类型的结构变异也更加完善[9-23]。CNV的识别也已从最初基于aCHG芯片的粗放式比较基因组杂交技术,发展为当前主流的基于测序数据序列覆盖度的统计分析检测技术。
当前对CNV识别的研究主要集中于对单一个体相对于参考序列的CNV识别和对两样本相对CNV的识别。随着高通量测序技术的进步、测序成本和生物信息学分析成本的下降,基于家系的三样本CNV识别成为可能且需求越来越大。相对于传统且成熟的配对数据CNV分析,基于家系CNV分析的优点在于可以同时得到一个家系中子女相对于其父母的新生CNV变异(denovo CNV)和继承自父母的变异(inherited CNV),而目前基于家系的CNV分析无论是识别工具还是案例分析都少有研究。
1 材料和方法 1.1 家系数据的获取及比对用于分析的家系基因组数据为来自千人基因组计划(http://www.1000genomes.org)的样本NA12878(母亲)、NA12877(父亲)及NA12880(女儿)。3个样本的数据均为使用HiSeq® 2000测序仪测得50X覆盖度的高通量测序数据。在得到样本的FASTQ格式Pair-End序列后,采用BWA-0.7.5a [24]设bwtsw参数进行比对。
1.2 基于家系数据的CNV识别基于BWA序列比对得到NA12878、NA2877及NA12880的SAM文件之后,根据其中双末端测序短序列(read pairs)的映射位置、SAM FLAG值以及粗粒化处理后的CIGAR值,按染色体选取其中完全映射到参考序列且两端映射方向正常的read pairs计算步长为100 bp的滑窗覆盖度(sliding read depth)。对于每个染色体上三个样本的滑窗覆盖度序列,考虑到其中可能的由于测序不均匀以及比对软件的错误比对造成的影响,首先采用基于haar变换基的小波变换(wavelettransformation)对滑窗覆盖度序列进行降噪处理。基于三样本降噪后的滑窗覆盖度序列,根据修正后的区域滑窗reads数目的水平变化量得到可能的CNV候选。针对这些候选CNV,首先采用非参数秩和检验(mannwhitney u test)判断候选CNV区域的覆盖度相对两侧(flanking regions)水平变化的显著程度。考虑到此时涉及大量CNV候选覆盖度变化的检验,为了从整体上控制每个染色体上CNV识别的第一类错误概率,在所有CNV候选的非参数秩和检验结果基础上,采用Bonferroni多重检验进行结果修正,从而确保对每个染色体而言,其所有检验结果显著的CNV的整体第一类错误概率(family-wise error rate)得到有效控制。
2 结果与分析 2.1 家系数据CNV的聚类分析及成因解释在得到家系数据的的CNV后,为了初步了解这些CNV的形成,首先对每个家系CNV根据家庭中得三个样本CNV区域测序覆盖度相对于全基因组平均覆盖度的得失情况(gain and loss)进行标准化,得到衡量覆盖度得失的三维取值连续区间的向量,分别对应样本NA12878、NA12877、NA12880在该CNV区域的相对覆盖度变化情况。在得到基于所有家系CNV的相对覆盖度变化向量构成的矩阵后,通过对其进行使用L1个体距离及完全组间距离的分层聚类分析对所有CNV聚类,结果见图 1。

|
图 1 家系CNV的聚类分析及聚类各组中CNV区域家系三样本相对覆盖度分布箱线图 Figure 1 Hierarchical clustering of familial CNVs and related boxplot of normalized read depth of CNV regions for each cluster. In the upper subfigure, L1 distance-based hierarchical clustering analysis with complete linkage was performed on the normalized read depth of family members. Ten groups were generated by separating the clustering dendrogram at 0.75 with related heatmap of normalized family read depths. The lower subfigure shows boxplots of the distribution of normalized read depth within each group. |
以组间距离为0.75为分界线,将所有组间距离小于1的聚类分支视为1类,聚类结果中的CNV便被分成10组,并按照聚类结果中的顺序从左到右依次标记为1-10组。通过热图以及各组内CNV区域3个样本相对覆盖度变化的箱线图可以看到,10个组中CNV在父(NA12877)、母(NA12878)、子女(NA12880)这3个样本相对覆盖度变化的分布上都分别具有明显的特征。对每个家系CNV在一个样本上如果其CNV区域的相对覆盖度变化是1,即该样本上CNV区域的覆盖度与基因组平均覆盖度持平,则认为该样本CNV区域的基因型是纯合的AA型。如果某样本上CNV区域的相对覆盖度变化是0.5,即该样本上CNV区域的覆盖度接近基因组平均覆盖度的一半,则认为该样本CNV区域的基因型是杂合的Aa型。类似地,若某样本上CNV区域的相对覆盖度变化接近0,亦即该样本在CNV区域几乎没有reads覆盖,则推断该样本在CNV区域的基因型是纯合的aa型。根据遗传学中等位基因的概念,可以以此通过不同的变异形成方式解释各个组的家系CNV。
对于第1组(聚类结果标记为深红色)中的家系CNV,由图 1中的热图和箱线图可知,该组中家系CNV的平均基因型为父(NA12877)AA型,母(NA12878)aa型,子女(NA12880)继承为Aa型。同理,第2组中的家系CNV的形成为父(NA12877)Aa型,母(NA12878)aa型,子女继承为Aa型。类似地,其他8组家系CNV也可以根据热图和箱线图中在父、母、子女三样本上相对覆盖度变化结合等位基因的概念解释子女CNV的形成,见表 1。
| 表 1 10类家系CNV汇总 Table 1 Summary of the 10 groups of familial CNVs |
为了更为直观地展示每一组家系CNV的独特特征,本文从每组中各选取有代表性的家系CNV实例。通过直接将母(NA12878)、父(NA12877)、子女(NA12880)3个样本在各组代表性的CNV区域附近实际reads覆盖度序列的变化情况通过红、蓝、黑三色分别标注,绘成折线图直观地展示CNV区域内父、母、子女3个样本的reads覆盖度相对于两侧未变异区域的特点与变化。值得注意的是,聚类结果中的第8组和第10组。从箱线图和热图中可以看出,第10组以母AA型、父aa型、子女Aa型为主,但由于该类CNV与母Aa型、父aa型、子女Aa型的聚类距离非常近,后者由于数量较少被并入第10组CNV中。同样的,第8组以父Aa型、母AA型、子女AA型为主,但由于该类CNV与父Aa型、母Aa型、子女AA型的聚类距离非常近,后者同样因数量较少被并入第8组CNV中。因此在列举这两组的代表性CNV时,也增列被并入8、10组的这两种CNV,结果见表 2。
| 表 2 各类家系CNV示例 Table 2 Demonstration of familial CNVs within each group. Format of the second column is mother_allele_type (NA12878) | father_allele_type (NA12877) -> offspring_allele_type (NA12880). |
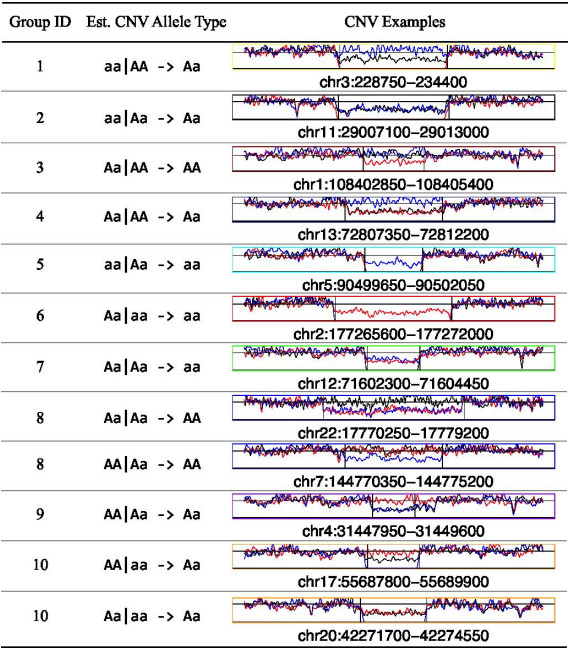
在前述不同生成模式的家系继承继承式CNV中,注意到其中第5、6及7组中后代的基因型均为aa型。与其他类继承式家系CNV不同,对于子女为纯合的aa型的家系CNV,如果在这些CNV区域存在exon,gene等,则在子女的基因组上则会出现由于缺失整段位于CNV区域的序列而与父、母中至少一方存在由缺少相应exon或gene所导致的表达上的差异。这可能正是这3组CNV的总数相对于其他组家系CNV数量偏少的原因之一。特别是对于聚类结果中的第7组中的继承式家系CNV,在CNV区域父、母的基因型都为显性的Aa型,这意味如果CNV区域存在exon或gene,则子女在表达上将存在于父、母均不一致的情况。因此,对于针对基因组上的结构变异特别是CNV对家庭后代所造成的病理影响的研究,则第5、6、7组特别是第7组中的家系CNV应该是这些研究的首要注意对象,这些家系CNV对家庭后代所造成的表型差异很有可能远大于其他CNV甚至其他结构变异的影响。图 2中列举了第7组中的所有23个家系CNV其变异区域附近各家庭成员的真实序列覆盖度情况。

|
图 2 第7组中所有家系CNV区域家庭成员的实际序列覆盖度情况 Figure 2 Read depth of family members near each familial CNVs in group 7. The allele types of the parents (colored by blue and red for father and mother, respectively) are both Aa, whereas that of the offspring (black) is aa. |
对于基于三样本或多子女样本的家系数据CNV分析,与传统的配对(paired datasets)CNV分析的不同之处除了上述对于家系继承式CNV产生方式的分析外,部分子女也可能会有少量相对于父、母的新生突变产生的CNV。然而,相对于继承得来得家系CNV而言,子女新生CNV的识别更具挑战。首先,现有研究显示这种新生突变产生的CNV数量极少[25-27],并非每一个子女都有,几乎没有可靠的训练集来进行针对新生CNV识别的优化;再者,广义上讲,任何由于突变产生而非父、母染色体搭配组合产生的相对于父、母染色体的差异都属于子女新生变异,然而由于在识别CNV时无法确认变异产生的原因,因此在识别子女新生CNV时,只有父、母双方某区段测序覆盖度均一致且与全基因组平均覆盖度相当,而子女的区域覆盖度出现显著降低时,这种CNV才会被判定为子女新生变异。在NA12878-NA12877-NA12880这一三样本家系数据中,经过分析和筛选认为较为可信的3个疑似子女新生CNV,在这些疑似子女新生CNV的发生区域,父(NA12877)、母(NA12878)双方的测序覆盖度折线均与基因组平均水平相当,而子女的测序覆盖度折线相对于父、母则显著下降,倘若CNV区域没有发生子女新生变异,则子女的区域测序覆盖度应当与父、母类似。
3 讨论相对于较为成熟的单一个体及两样本相对结构变异的检测,目前针对家系结构变异识别的研究尚处于起步阶段,而现有针对家系数据的识别工具也多集中于对家系SNP(single-nucleotide polymorphism)的识别,如TrioDeNovo [28]等。本文构建了基于小波变换和非参数统计检验的家系CNV识别方法,同时基于NA12878-NA12877-NA12880这一真实家系数据,本文详述了对子女继承式家系CNV的类型及生成方式的分析。在子女继承式CNV的聚类分析及生成方式的推断中,我们根据对家系数据的相对覆盖度变化矩阵的分类结果,结合遗传学中等位基因的概念可以很好地解释各个组绝大多数子女CNV的继承得来方式。这种推断并不是对所有的家系CNV都准确、可靠。
在对子女继承式家系CNV的分析中,我们注意到有小部分家系CNV的推断存在一定程度的偏差。这主要是由于对CNV的分组和推断都是基于原始的家系数据中CNV区域的测序覆盖度序列,使得分析的可靠性直接依赖于CNV区域测序覆盖度的计算的准确性。然而基于当前的测序及比对技术,有众多因素都会造成后期覆盖度计算的偏差。例如测序过程中基因组区域抽样的不均一性,序列比对时由于基因组高重复度区域等的复杂性及比对软件缺陷等造成的序列错误映射等,都会对区域覆盖度的计算造成系统性的偏差。虽然大多数情况下,这些偏差造成的影响较小,相对于区域覆盖度的信号强度只产生微弱的干扰。体现在前文的分析中即图 1箱线图中大多数组里所体现的相对于均值在正常范围内的浮动,但除了前文中提到的由于聚类距离过近造成的第8、10组将两类不同生成模式的CNV聚在一起外,第4组家系CNV在相对覆盖度向量的三个坐标上的方差也显著且非正常地高于其他组。这一方面是由于第4组CNV的数量多于其他组使得组内的差别变大,但本文推断其主要原因应该为在子女数据(NA12880)的CNV区域由于测序不均一或序列的错误映射造成了区域序列覆盖度系统性的偏少。
除了由于原始数据中区域序列覆盖度的系统性偏差造成的影响外,对覆盖度的标准化过程也有可能对少数位于高覆盖度区域的家系CNV的分析造成影响。对于家系CNV,如果在父、母、子女三个样本中CNV区域的原始序列覆盖度向量为〈 cf, cm, co 〉,则归一化得到相对覆盖度变化向量的计算方式为
| $ \begin{gathered} \left\langle {\frac{{{c_f}}}{{\max \left( {{c_f}, {c_m}, {c_o}, {c_{avg}}} \right)}}, \frac{{{c_m}}}{{\max \left( {{c_f}, {c_m}, {c_o}, {c_{avg}}} \right)}}, } \right. \hfill \\ \left. {\frac{{{c_o}}}{{\max \left( {{c_f}, {c_m}, {c_o}, {c_{avg}}} \right)}}} \right\rangle, \hfill \\ \end{gathered} $ |
其中cavg为基因组的平均序列覆盖度。对于绝大多数家系CNV而言,这种计算方式简单有效,但对于本身位于高覆盖度区域的家系CNV而言,就会产生误导性的偏差。例如,一个位于平均覆盖度2倍于基因组平均水平区域的CNV,如果在三样本家系数据中,父母一方和子女由于为纯合(aa型)而局部覆盖度为0,另一方由于为杂合(Aa型)而局部覆盖度接近基因组平均水平时,根据上述归一化方法,则会将杂合Aa型的父/母错误归类为显性纯合(AA型)。
在对家系继承CNV进行分组及生成方式的推断时,我们注意到了不同组之间CNV数量相对于孟德尔遗传定律的反常。例如生成方式对称的第5组和第6组,从遗传学和概率上讲,理应数量相当,但在NA12878-NA12877-NA12880的家系数据中却出现了反常,其原因可能在于现有的CNV识别算法对于不同配对样本的识别存在精度不一致的现象。在分别进行子女相对于父、母一方的两样本配对CNV分析时我们发现,子女(NA12880)相对于母方(NA12878)的CNV数量显著少于相对于父方(NA12877)的CNV数量,这就导致在大多数的继承方式对称的组如第5组相对于第6组、第4组相对于第9组中,子女与父方(NA12877)存在差异的组中CNV的数量要显著大于子女与母方(NA12878)存在差异的组。我们发现除了本文方法之外,使用其他的CNV识别方法[10, 19, 23]以及针对不同的家系数据,都存在类似的子女相对于父、母CNV数量的差异。这些结果说明现有CNV识别方法在从配对数据的CNV推广到基于多样本的家系CNV识别时,可能存在一定的缺陷而导致结果存在偏差,需要建立更为全面和准确的基于谱系数据的CNV识别算法和工具。
| [1] | Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping[J]. Nat Rev Genet,2011 (12) : 363-76. |
| [2] | Feuk L, Carson AR, Scherer SW. Structural variation in the human genome[J]. Nat Rev Genet,2006 (7) : 85-97. |
| [3] | Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing[J]. Nat Methods,2009, 6 (11, S) : S13-20. |
| [4] | Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease[J]. Annu Rev Med,2010 (61) : 437-55. |
| [5] | Almal SH, Padh H. Implications of gene copy-number variation in health and diseases[J]. J Hum Genet,2012, 57 (1) : 6-13. DOI: 10.1038/jhg.2011.108. |
| [6] | Bassett AS, Scherer SW, Brzustowicz LM. Copy number variations in schizophrenia:critical review and new perspectives on concepts of genetics and disease[J]. Am J Psychiatry,2010, 167 (8) : 899-914. DOI: 10.1176/appi.ajp.2009.09071016. |
| [7] | Ionita-Laza I, Rogers AJ, Lange CA, et al. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis[J]. Genomics,2009, 93 (1) : 22-6. DOI: 10.1016/j.ygeno.2008.08.012. |
| [8] | Zhang F, Gu WL, Hurles ME, et al. Copy number variation in human health, disease, and evolution[J]. Annu Rev Genomics Hum Genet,2009, 10 (10) : 451-81. |
| [9] | Abyzov A, Urban AE, Snyder M, et al. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing[J]. Genome Res,2011, 21 (6) : 974-84. DOI: 10.1101/gr.114876.110. |
| [10] | Boeva V, Zinovyev A, Bleakley K, et al. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization[J]. Bioinformatics,2011, 27 (2) : 268-9. DOI: 10.1093/bioinformatics/btq635. |
| [11] | Chiang DY, Getz G, Jaffe DB, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing[J]. Nat Methods,2009 (6) : 99-103. |
| [12] | Duan J, Zhang JG, Deng HW, et al. Comparative studies of copy number variation detection methods for next-generation sequencing technologies[J]. PloS one,2013, 8 : e59128. DOI: 10.1371/journal.pone.0059128. |
| [13] | Ivakhno S, Royce T, Cox AJ, et al. CNAseg-a novel framework for identification of copy number changes in cancer from secondgeneration sequencing data[J]. Bioinformatics,2010, 26 (24) : 3051-8. DOI: 10.1093/bioinformatics/btq587. |
| [14] | Teo SM, Pawitan Y, Ku CS, et al. Statistical challenges associated with detecting copy number variations with next-generation sequencing[J]. Bioinformatics,2012, 28 (21) : 2711-8. DOI: 10.1093/bioinformatics/bts535. |
| [15] | Yoon S1, Xuan Z, Makarov V, et al. Sensitive and accurate detection of copy number variants using read depth of coverage[J]. Genome Res,2009, 19 (9) : 1586-92. DOI: 10.1101/gr.092981.109. |
| [16] | Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing[J]. Nat Genet,2009, 41 (10) : 1U29-061. |
| [17] | Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing[J]. BMC Bioinformatics,2009, 10 : 80. DOI: 10.1186/1471-2105-10-80. |
| [18] | Simpson JT, Mcintyre RE, Adams DJ. Copy number variant detection in inbred strains from short read sequence data[J]. Bioinformatics,2010, 26 (4) : 565-7. DOI: 10.1093/bioinformatics/btp693. |
| [19] | Medvedev P, Fiume M, Dzamba M, et al. Detecting copy number variation with mated short reads[J]. Genome Res,2010, 20 (11) : 1613-22. DOI: 10.1101/gr.106344.110. |
| [20] | Waszak SM, Hasin Y, Zichner T, et al. Systematic inference of copy-number genotypes from personal genome sequencing data reveals extensive olfactory receptor gene content diversity[J]. PLoS Comput Biol,2010, 6 (11) : e 1000988. DOI: 10.1371/journal.pcbi.1000988. |
| [21] | Kim TM, Luquette LJ, Xi R, et al. rSW-seq: algorithm for detection of copy number alterations in deep sequencing data[J]. BMC Bioinformatics,2010, 11 : 432. DOI: 10.1186/1471-2105-11-432. |
| [22] | Duan J, Zhang JG, Lefante J, et al. Detection of copy number variation from next Generation sequencing data with total variation penalized least square optimization[C] //In Bioinformatics and Biomedicine Workshops, 2011: 3-12. |
| [23] | Xi R, Luquette J, Hadjipanayis A, et al. BIC-seq: a fast algorithm for detection of copy number alterations based on high-throughput sequencing data[J]. Genome Biol,2010, 11 : O10. DOI: 10.1186/1465-6906-11-S1-O10. |
| [24] | Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform[J]. Bioinformatics,2009, 25 : 1754-60. DOI: 10.1093/bioinformatics/btp324. |
| [25] | Itsara A, Wu H, Smith JD, et al. De novo rates and selection of large copy number variation[J]. Genome Res,2010, 20 (11) : 1469-81. DOI: 10.1101/gr.107680.110. |
| [26] | Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism[J]. Science,2007, 316 (5823) : 445-9. DOI: 10.1126/science.1138659. |
| [27] | Xu B, Roos JL, Levy S, et al. Strong association of de novo copy number mutations with sporadic schizophrenia[J]. Nat Genet,2008 (40) : 880-5. |
| [28] | Wei Q, Zhan X, Zhong X, et al. A bayesian framework for de novo mutation calling in parents-offspring trios[J]. Bioinformatics,2015, 31 (9) : 1375-81. DOI: 10.1093/bioinformatics/btu839. |